Warning: Undefined variable $PHP_SELF in /customers/9/8/1/usmart.dk/httpd.www/bachelor/index.php on line 2
Warning: Undefined array key "word" in /customers/9/8/1/usmart.dk/httpd.www/bachelor/index.php on line 5
Warning: Undefined array key "svar" in /customers/9/8/1/usmart.dk/httpd.www/bachelor/index.php on line 6
Warning: Undefined array key "valg" in /customers/9/8/1/usmart.dk/httpd.www/bachelor/index.php on line 7
Spanners
Structural biology is the branch of molecular biology concerned with the architecture and shape of proteins. Proteins are one of the basic building materials of all organic matter, taking shape as enzymes, antibody units used by the immune defense, or performing a wide array of other biological tasks.
Proteins fold into certain shapes, depending on its structure and on the surroundings, and the way it folds effect which properties the protein posesses. Therefore proteins, and not least the protein folding process, are of vivid interest to biologists.
Luckily, t-spanners may provide a valuable tool in this research.
The structure of a Protein
Without getting into too much detail, a protein consists of strings of amino acids connected by peptide bonds. The combination of amino acids, and the way in which they are arranged, is known as the primary structure of the protein.
The peptide bonds bend the chain of amino acids at an angle and have some flexibility, allowing the protein to rotate around the bonds. In this way the protein can fold into different shapes. The shape created by this folding is called the tertiary structure of the protein.
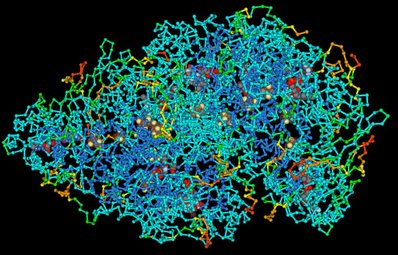
Figure 12: The tertiary structure of a protein
Proteins can be quite large and consist of tens of thousands of connected amino acids.
Photo from Wikipedia (http://en.wikipedia.org/wiki/Protein).
Protein folding
It appears that proteins with the same primary structure and in the same surroundings tend to fold into exactly the same tertiary structure. The process of folding takes from a few microseconds to several hours, depending on the surroundings and on the complexity of the protein.
The way in which a protein folds has influence on which properties it posseses. Under some conditions the protein can dissove and/or fold in an incorrect way. Incorrectly folded protein, prions, have been related to illnesses such as Creutzfeldt-Jakob disease and BSE (Mad cow disease).
This makes the study of protein folding a subject of massive interrest. Never the less, we are still not quite sure what happens during the folding process.
Modelling the protein
Because proteins can be quite large (the muscle protein titin is a chain of about 27.000 amino acids) keeping track of the individual parts of it during folding is quite difficult. One way is to make 3D-representations like the illustration above. However, it may be difficult to see what is going on.
One way of creating a better overview is to focus on fewer, more interesting areas of the protein. These interesting places are where amino acids come close to each other during the folding process.
A t-spanner constructed by the greedy algorithm has the property, that points located close to each other tend to be connected. A t-spanner constructed for a string of amino acids will, with an appropriate value of t, consist of edges along the spine of the protein as well as between amino acids which have come close to each other during the forling proces. If several protein states are recorded during the folding it will suffice to check which edges appear in the spanner and which are lost, to get an understanding of how the folding takes place.
The limited space needed to store the t-spanners (compared to storing all distances) and the relatively fast algorithm for construction t-spanners make it a good tool in the studies of protein folding.
More information on modelling of proteins is available in the further reading section.
An exercise on protein folding is available in the exercises section.